Stereo-seq Mouse Embryo Data Application
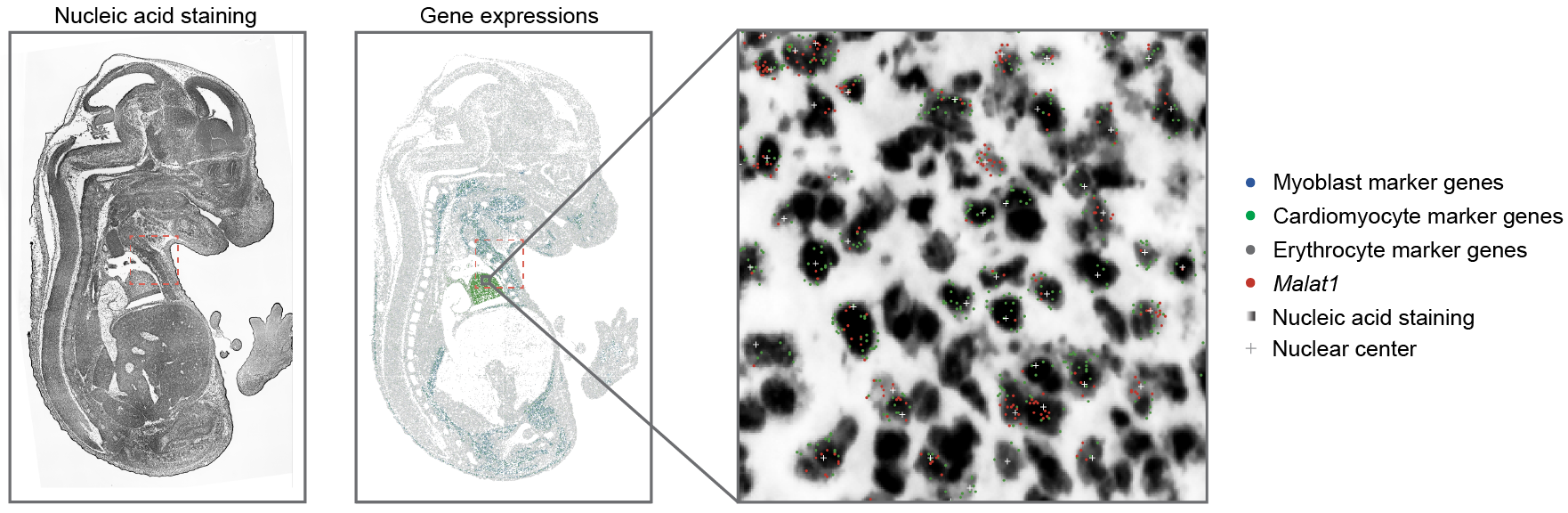
For each cell type, we first preprocess the input data
python -m ella.data.prepare_data -i your_dir/data_Myoblasts.pkl -o prepared_data
Then run ELLA across genes in parallel for each cell type with a corresponding recipe (.yaml file)
bash run_stereoseq.sh
Other scripts used for mRNA characteristic analysis and for plotting are shared in the github repo.