A Complete Demo
Here’s a demo introducing a relatively complete ELLA analysis pipeline.
The script and data that will be used in this demo should have already been downloaded (while cloning the ELLA repo). You should be able to find these at your local ELLA folder:
ELLA/tutorials/complete_demo/
├── input
│ └── complete_demo_data.pkl
├── output
│ ├── df_nhpp_prepared.pkl
│ ├── df_registered.pkl
│ ├── lam_est.pkl
│ ├── nhpp_fit_results.pkl
│ └── pv_est.pkl
└── complete_demo.ipynb
The data is a subset of the processed seqFISH+ embryonic fibroblast dataset. The input data (input/complete_demo_data.pkl) mainly contains a dictionary of three dataframes corresponding to gene expression, cell segmentation, and nucleus segmentation (optional) with 20 cells and 50 genes.
The script of this demo is complete_demo.ipynb, you should be able to run it locally by yourself (run time around 3min), expecting the following steps and outputs.
The alternative NHPP fit uses a bounded-Newton solver (deterministic, finds the global optimum); there are no adam_* or max_iter arguments.
- Initiating ELLA:
# import ELLA from ELLA.ELLA import ELLA ella_demo = ELLA(dataset='demo') # load data ella_demo.load_data(data_path='input/complete_demo_data.pkl') - Run the data pre-processing and model fitting steps:
# register cells ella_demo.register_cells() # prepare data for model fitting ella_demo.nhpp_prepare() # model fitting ella_demo.nhpp_fit()The bounded-Newton fit is fast (this 50-gene / 20-cell demo fits in ~1-2 s), so we just fit live above. For large panels you can instead save the fit and reload it later with:
# load a previously saved fit ella_demo.load_nhpp_fit_results(res_path='output/nhpp_fit_results.pkl') - Let’s then run the testing and estimation:
# expression intensity estimation ella_demo.weighted_density_est() # likelihood ratio test ella_demo.compute_pv() - We can now cluster the estimated (significant) expression intensities into clusters of patterns. We find the optimal number of kmeans clusters K with the ELBOW method where K is chosen as a point where the distortion/inertia begins to decrease more slowly:
ella_demo.pattern_clustering()
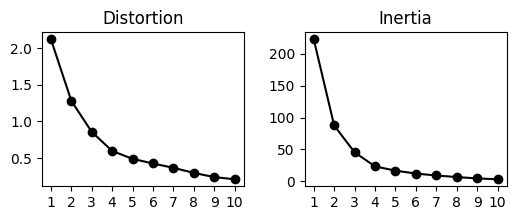
Based on the plots, it seems 5 can be a proper choice, thus let’s proceed with K=5 to obtain 5 pattern clusters:
ella_demo.pattern_labeling(K=5)
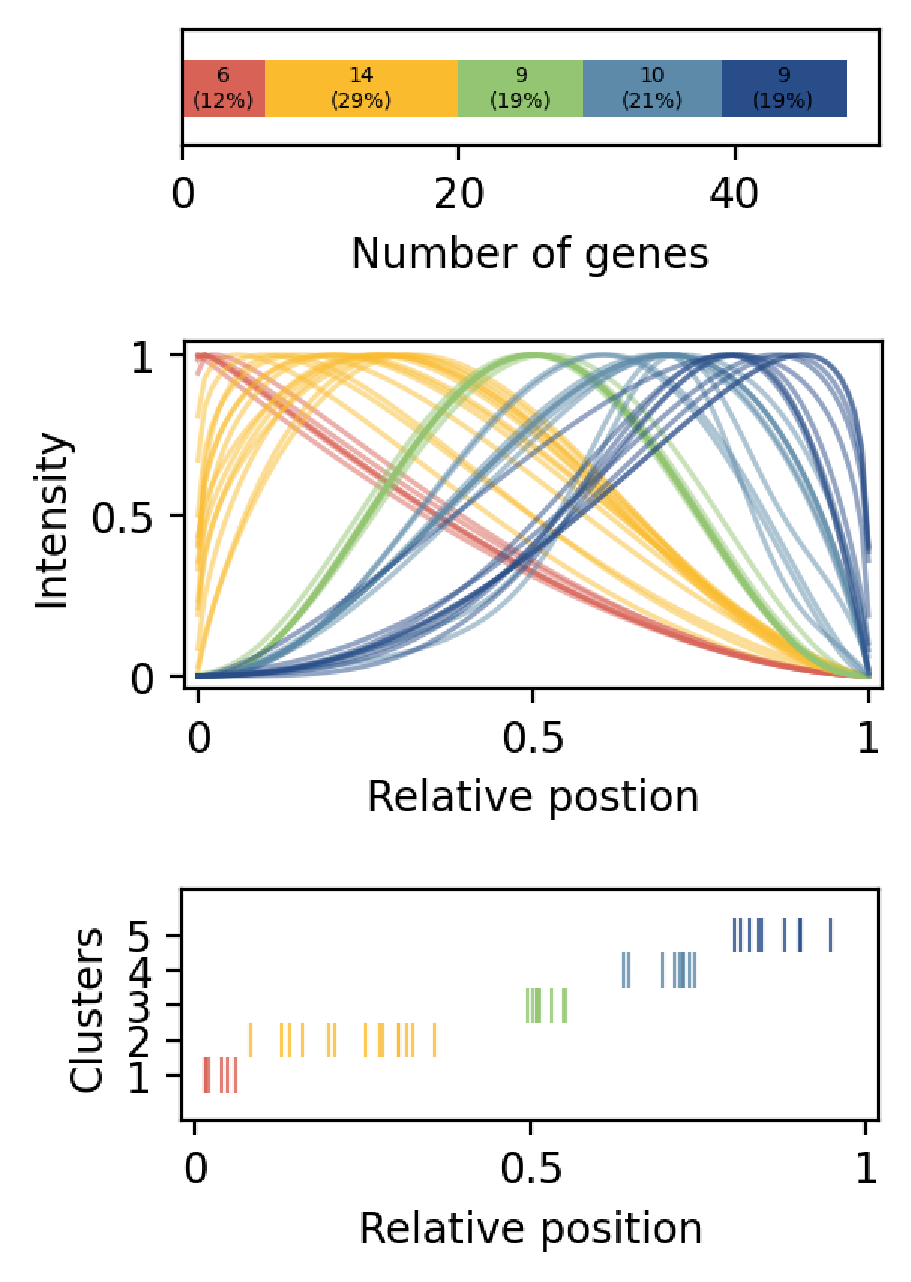
Prints:
Pattern 1: 5 genes
Pattern 2: 14 genes
Pattern 3: 11 genes
Pattern 4: 9 genes
Pattern 5: 6 genes
Plots: numbers and proportions of significant genes, estimated expression patterns, and estimated pattern scores
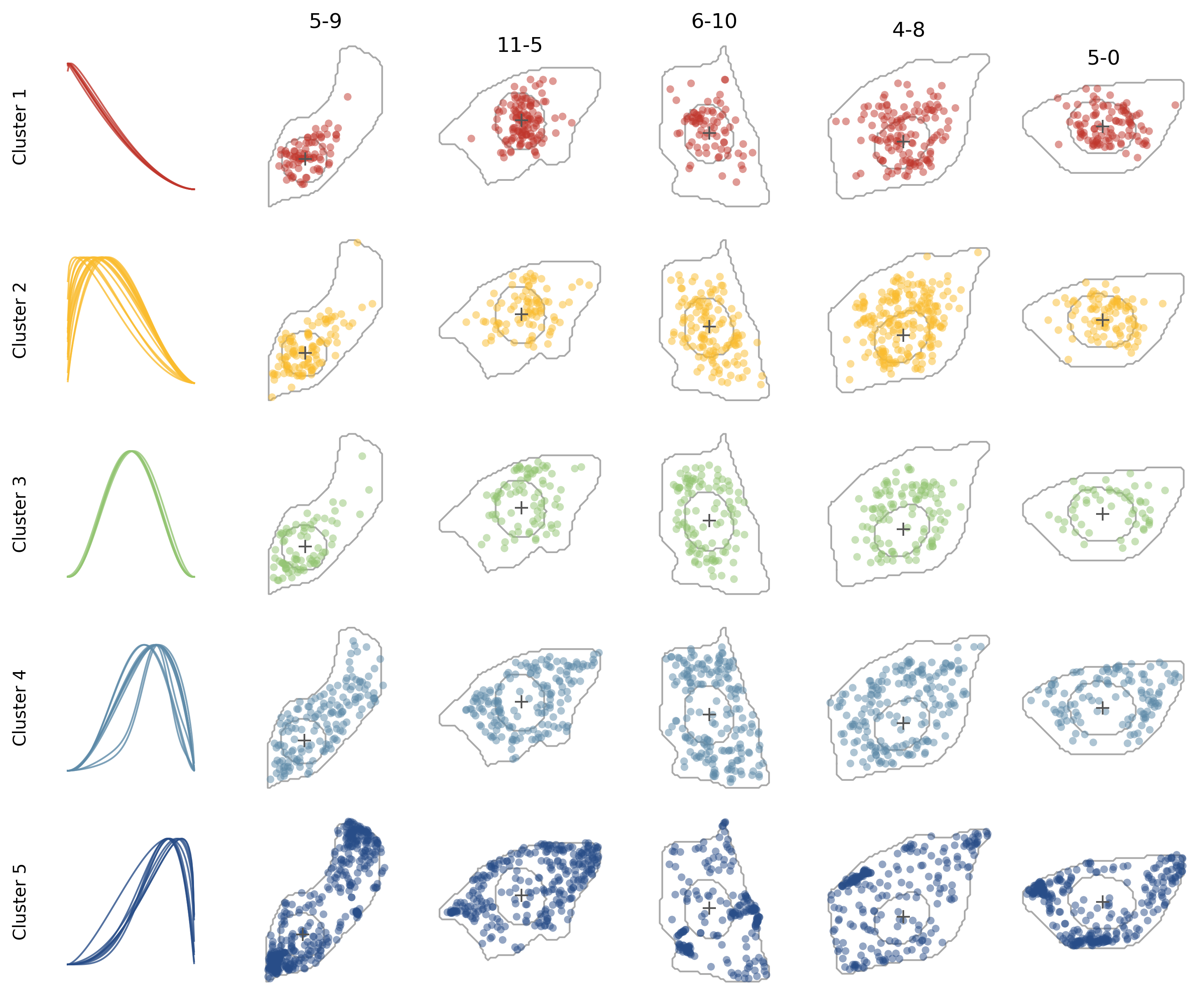
We can overlay all genes in the same cluster in cells to have a more intuitive sense of the patterns.