Inputs
ELLA takes a dictionary of inputs. The two main pieces are a gene expression table (expr) and the cell boundary polygons (cell_poly).
1. Gene expression with nuclear center
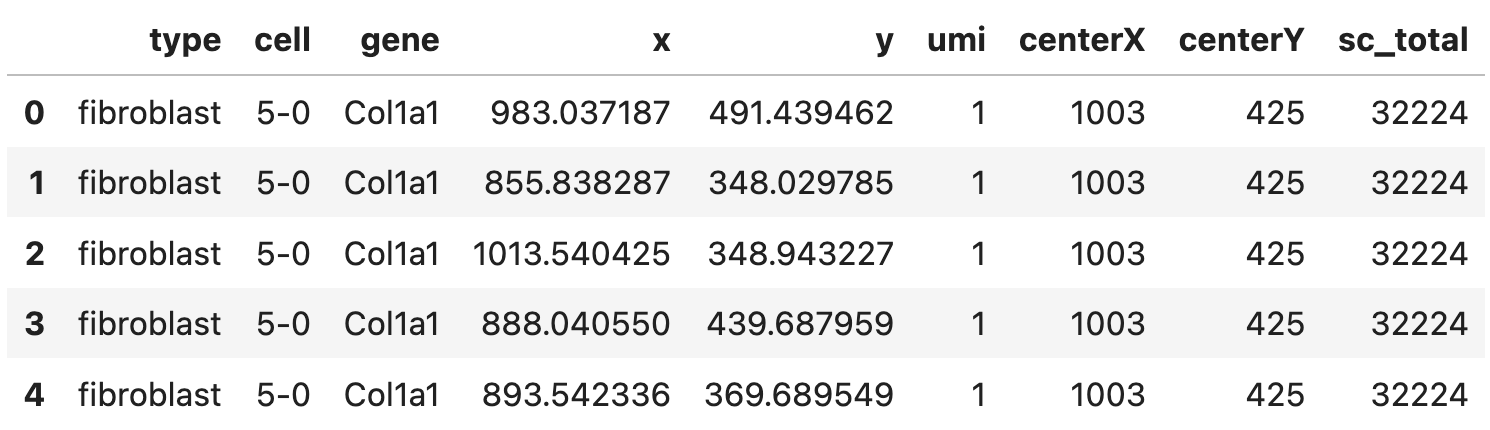
A pandas data frame (expr) with a few columns:
- spatial gene expression including the coordinates (
x,y) and the corresponding counts (umi) - cell center (
centerX,centerY) - cell type (
type), cell ID (cell), gene ID (gene) - and the total number expression counts of cells (
sc_total), rows corresponding to the same cell should have the same value for this column.
Here’s how the data frame looks like:
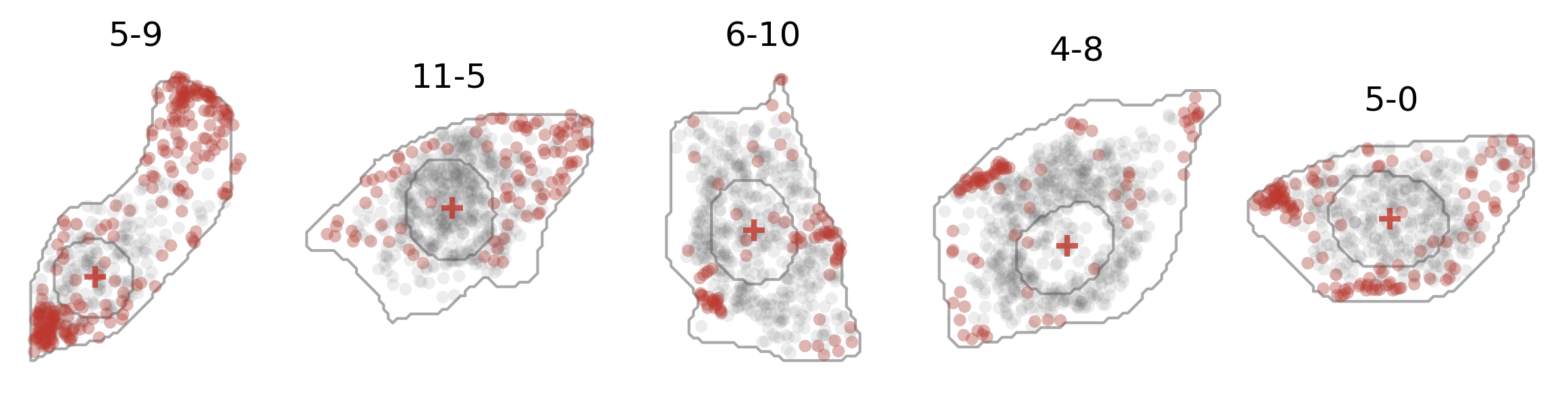
And here’s how the expression of one gene (dots in red) and the cell center (crosses in red) looks like:
2. Cell boundary polygons
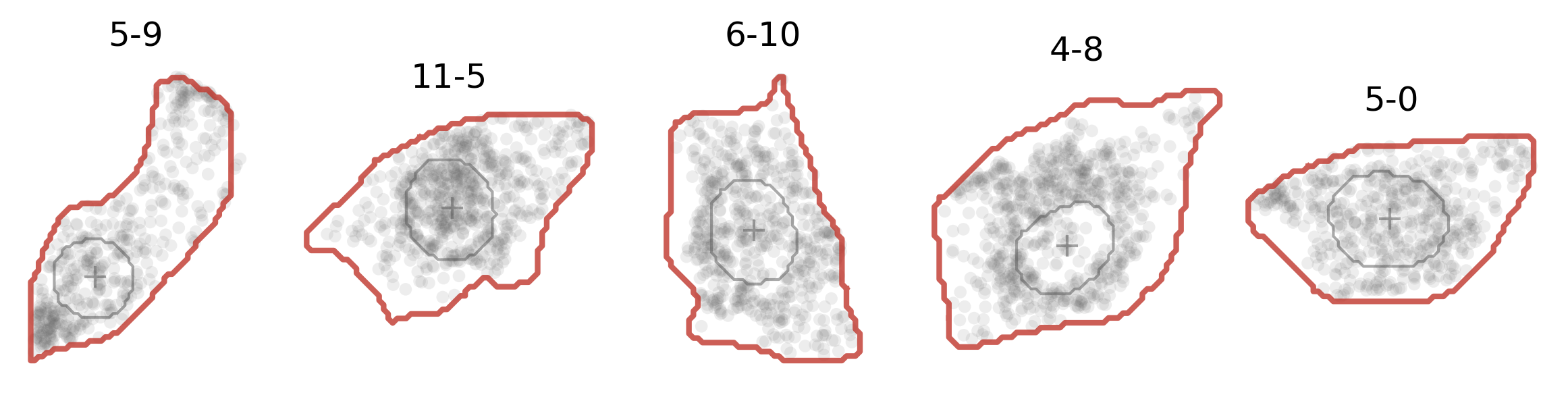
A dictionary (cell_poly) mapping each cell ID to its boundary polygon: an (M, 2) array of (x, y) vertices in the native coordinate frame. ELLA registers each cell to the unit disk by ray-casting against this polygon, so a polygon is required for every cell.
Here’s how the cell boundary looks (in red solid line):
A raster cell segmentation point cloud (cell_seg: a data frame of boundary (cell, x, y) points) is optional and used only for visualization, for example drawing the outlines in the demos; ELLA itself no longer consumes it. It looks like:
Nucleus segmentation
[Optional, for visualization purpose ONLY]
A pands dataframe (nucleus_seg) with 3 columns:
- cell ID (
cell) - the coordinates of points that characterize the nucleus segmentation boundary (
nucleus_seg).
Here’s how the data frame looks like:
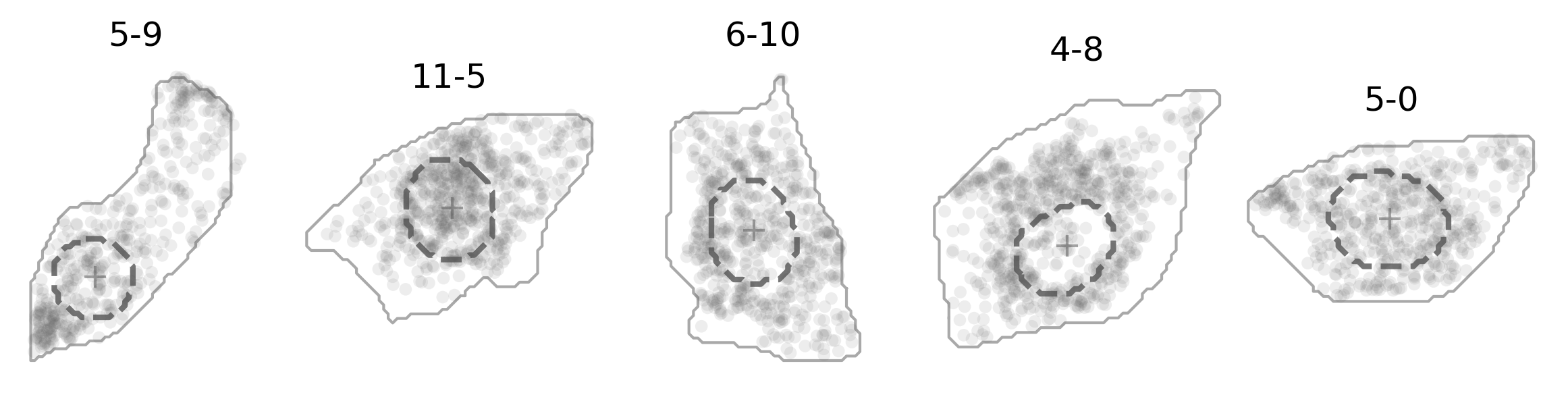
And here’s how it actually looks like (in darkgray dashed line):
Other required inputs
typesa list corresponding to all cell types.cellsa dictionary of lists corresponding to list of cells in each cell type.cells_alla list of all cells across cell types.genesa dictionary of lists corresponding to list of genes in each cell type.
How about tweak your own data into the format that ELLA takes and have a try!
Outputs
Running the pipeline (register_cells → nhpp_prepare → nhpp_fit → weighted_density_est → compute_pv, and optionally pattern_clustering → pattern_labeling) stores the results on the ELLA object as dictionaries keyed by cell type. The main ones (these are what the demos read):
- Significance (
pv_fdr_tl[type]): per-gene FDR-adjusted p-values (Benjamini-Yekutieli). A gene is significant atsig_cutoff(default 0.05), meaning it has a non-uniform subcellular localization pattern. The pre-adjustment p-values are also kept:pv_cauchy_tl(Cauchy-combined across the 22 kernels) andpv_raw_tl(per-kernel), with the likelihood-ratio test statistics ints_tl. - Estimated intensity (
weighted_lam_est[type]): per gene, the model-averaged expression-intensity curve as a function of relative position (0 = nuclear center, 1 = cell membrane), evaluated on a length-100 grid. This is the curve the demos plot. - Peak score (
scores[type]): per gene, the relative position where the estimated intensity peaks, a scalar summary of where expression concentrates. - Pattern clusters (
labels_dict[type]): afterpattern_labeling(K), the k-means cluster label per gene, with clusters ordered by peak position; non-significant genes are left unlabeled.
Lower-level fit results are available too: the fitted parameters and max log-likelihood per gene and kernel (A_est, B_est, mll_est), the model-averaging weights (weight_ml), and the best kernel per gene (best_kernel_tl).
Each stage also writes a pickle to output/, so results can be reloaded without refitting:
| file | written by | contents |
|---|---|---|
df_registered.pkl | register_cells | cells registered to the unit disk |
df_nhpp_prepared.pkl | nhpp_prepare | per-gene data prepared for fitting |
nhpp_fit_results.pkl | nhpp_fit | fitted A_est, B_est, mll_est |
lam_est.pkl | weighted_density_est | scores, weights, weighted_lam |
pv_est.pkl | compute_pv | FDR-adjusted p-values |
See A Complete Demo for how these outputs are produced and used end-to-end.